Makale
Biyobenzer İlaçların Yasal Mevzuatları
11 Ocak 2021, Pt
Sağlık sektörünün sacayakları olan medikal teknolojiler, ilaç ve biyoteknoloji alanlarında Avrupa Patent Ofisi’ne (EPO) 2019 yılında yapılan patent başvuruları, tüm alanlar içerisinde ilk onda yer almaktadır.

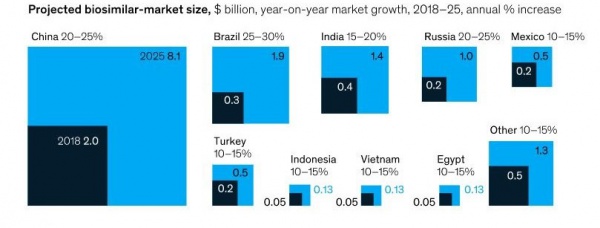
Biyolojik ürünler, global sağlık otoritelerince farklı tanımlara sahip olmakla birlikte, mikroorganizma, bitki hücresi veya hayvan hücresi gibi canlı bir sistemde biyoteknoloji yoluyla üretilebilen ve karakterize edilmesi genellikle küçük moleküllü ilaçlardan daha zor olan genellikle büyük, karmaşık moleküllerdir. Günümüzde sağlık hizmetlerine erişimin artırılmasına yönelik global odaklanma ve bilim ve teknoloji alanındaki gelişmelerle birlikte biyolojik ilaç pazarının büyük kısmını oluşturan patentlerin yakın zamanda sona erecek olmasının da etkisiyle, dünyada ve ülkemizde biyoteknolojik ilaç pazarı giderek büyümektedir. 2018 yılında yaklaşık 2 milyar Amerikan Doları değerinde olan Çin biyobenzer pazarının 2025 yılına kadar yılda %20-25 büyüyerek 8 milyar Amerikan Doları sınırını aşması, 2018 yılında 200 milyon Amerikan Doları değerinde olan Türkiye biyobenzer pazarının ise yılda %10-15 büyüyerek 500 milyon Amerikan Doları değerine ulaşması öngörülmektedir.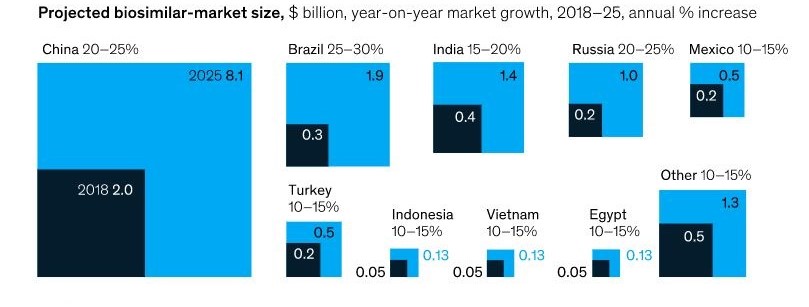
Jenerik ilaçlar, orijinal bir kimyasal ilaç etken maddesinin eşdeğeri veya birebir kopyası iken biyolojik kaynakların doğası gereği ve her üreticiye özgü üretim sürecinden dolayı, biyobenzer ve referans biyolojik ilaç arasında bazı farklılıklar oluşabilmektedir. Nitekim global sağlık otoritelerinin biyobenzer ve referans ürün tanımı, ruhsatlandırma süreci, eşdeğerlik ve isimlendirme yaklaşımı farklılık gösterebilmektedir.
Avrupa Birliği regülasyonlarında benzer biyolojik uygulamalar temelini 2001/83/AT sayılı Beşeri Tıbbi Ürünler Hakkındaki Avrupa Parlamentosu ve Konsey Direktifi ile 726/2004/AT sayılı Beşeri Tıbbi Ürünler İçin Koşullu Ruhsat Hakkındaki Avrupa Parlamentosu ve Konsey Tüzüğü’nden almakta olup ayrıca bu kapsamda pek çok rehber yayımlanmıştır. Bu kapsamda biyobenzer ilaç, Avrupa Ekonomik Alanı’nda piyasaya sürülmüş olan bir referans biyolojik ilaca benzer olduğu karşılaştırılabilirlik çalışmaları ile kanıtlanmış tıbbi ürün olarak tanımlanmaktadır. Biyolojik ilaçların doğal değişkenliğinin ilacın çalışma şeklini veya güvenliğini etkilememesini sağlamak için üretim aşamasında sıkı kontroller öngörülmüş olup bu sıkı kriterlerin benzer şekilde biyobenzer ilaçlara da uygulanmasını gerektiren regülasyonlar mevcuttur. Bu kapsamda biyobenzerlerin geliştirilmesi büyük ölçüde karşılaştırılabilirlik çalışmalarına dayanmaktadır ve referans ürün ile biyobenzer arasında nitelik, güvenlik ve etkililik açısından önemli bir fark olmaması aranmaktadır.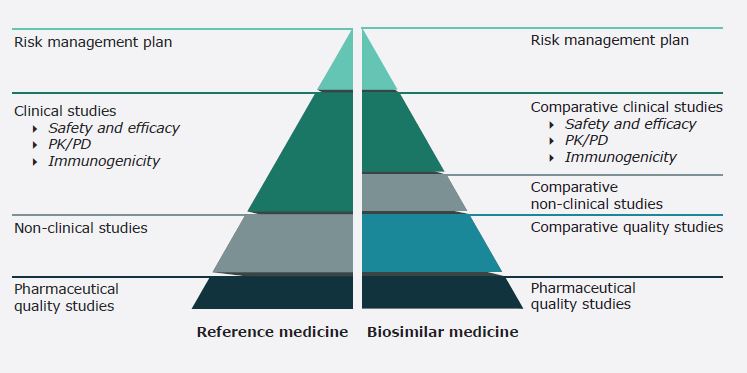
Biyobenzerlerin Avrupa Birliği’nde kullanım için piyasaya sürülmesi, son onayı Avrupa Birliği Komisyonu tarafından yapılmak suretiyle merkezi onay sistemine tabi olup kural olarak karşılaştırılabilirlik çalışmalarına Avrupa İlaç Dairesi (EMA) tarafından ruhsatlandırılmış tek bir referans ürün esas alınmaktaysa da klinik araştırmaların tekrarından kaçınmak için Avrupa Birliği dışında ruhsatlandırılmış bir ürünün referans alınması da söz konusu olabilmektedir. Avrupa Birliği ve EMA yaklaşımında değiştirilebilirlik, aynı klinik etkiye sahip olması beklenen bir ilacı başka bir ilaçla değiştirme olasılığını ifade etmekle bu, bir referans ürünü biyobenzerle (veya tam tersi) değiştirmek veya bir biyobenzerini diğeriyle değiştirmek anlamına gelebilmektedir. Biyobenzerler bakımından değiştirilebilirliğe izin verilmesi, ulusal düzeyle yapılacak regülasyona tabidir.
Dünya Sağlık Örgütü tarafından da biyolojik standardizasyon çalışmaları kapsamında Biyobenzerlerin Değerlendirilmesine İlişkin Rehber yayımlanmış olup benzer biyoterapötik ürün ruhsatlandırılmasına ilişkin kararları destekleyen kanıt standardının, ürünlerin halk sağlığı amaçları için kabul edilebilir kalite, güvenlik ve etkililik düzeylerinin karşılanması için yeterli olması gerekliliği vurgulanmıştır. Fikri mülkiyet sorunları, referans biyoterapötik ürün ile benzer biyoterapötik ürünün değiştirilebilirliği, etiketleme ve reçete bilgileri gibi hususların ise ulusal düzenleyici otoritelerce belirlenmesi gerektiği belirtilmiştir.
2009 tarihli Biyolojik Fiyat Rekabeti ve İnovasyon Yasası (BPCI Act) ile Amerika Birleşik Devletleri’nde biyobenzer olduğu veya Amerikan Gıda ve İlaç Dairesi (FDA) onaylı bir biyolojik ürünle değiştirilebilir olduğu ortaya koyulan biyolojik ürünler için kısaltılmış bir ruhsatlandırma yolu oluşturulmuştur. Bu kapsamda biyobenzer, referans biyolojik ürünle güvenlilik, saflık ve etkinlik açısından klinik olarak anlamlı farklılıkları olmadığı ortaya koyulan biyolojik ürün olarak tanımlanmaktadır. Referans ürünün ise güvenliğini ve etkililiğini göstermek için gerekli tüm verileri ve bilgileri içermesi gereken bağımsız bir başvuru ile FDA tarafından onaylanmış olması gerekmektedir. FDA tarafından onaylanmamış bir referans ürün esas alınması durumunda biyobenzer, FDA onaylı benzer referans ürün ve ABD dışında ruhsatlandırılmış referans ürün arasında üçlü karşılaştırmalı çalışma yapılması mümkündür. Karşılaştırmalı veriler, ürünlerin ayrıntılı analitik (yapısal ve işlevsel) karakterizasyonu ve karşılaştırması ile başlayan aşamalı bir şekilde oluşturulmakta ve değerlendirilmektedir (Görsel 4). Dolayısıyla, biyobenzerin FDA onaylı referans üründen klinik olarak anlamlı farklılıkları olmadığını gösteren bir üretici, referans ürünle aynı klinik olmayan ve klinik verilerin tam profilini oluşturmak yerine FDA’in referans ürün için gerçekleştirdiği güvenilirlik ve etkililik tespitine dayanabilecektir. Değiştirilebilir ürünler bakımından sağlanması gereken başkaca kriterlerin yanı sıra biyobenzerin herhangi bir hastada referans ürünle aynı klinik sonuçları üretmesinin beklendiği ortaya koyulmalıdır. Bu kapsamda, değiştirilebilir ürünlerin reçete eden yetkilinin inisiyatifi dışında birbirleriyle değiştirilmesi mümkündür. 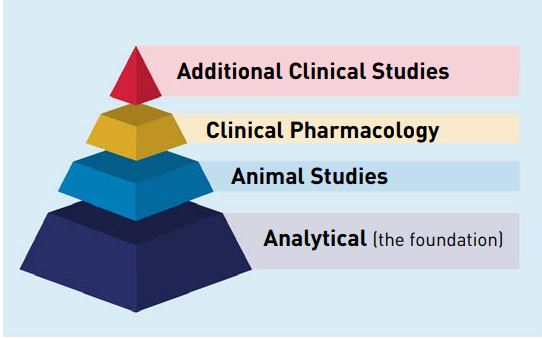
Çin Gıda ve İlaç Dairesi (“CFDA”) tarafından 2015 yılında biyobenzerlerin araştırılması, geliştirilmesi ve değerlendirilmesi için ilkeleri, referans ürünlerin kullanım yöntemini, farmasötik, klinik olmayan, klinik çalışmalar ile bunların değerlendirilme yöntemini ortaya koyan bir rehber yayımlamış olup yakın zamanda biyobenzerlere ilişkin yaygın olarak ele alınan referans ilaç, karşılaştırılabilirlik gibi konularda rehberler yayımlanmıştır. Buna göre biyobenzer, halihazırda CDFA tarafından ruhsatlandırılmış orijinal ürünle aynı aminoasit yapıya sahip olan ve kalite, güvenlik ve etkililik açısından benzer biyoterapötik ürün olarak tanımlanmaktadır. FDA ve EMA yaklaşımından farklı olarak CFDA dışında bir kurumca onaylanan biyolojik ürünlerin, yahut CFDA onaylı bir biyobenzerin ise tek başına referans ürün olarak kullanılması mümkün değildir. İlaveten, biyobenzerlerin ruhsatlandırılma başvuruları yeni ilaç başvuruları ile aynı yönteme tabi olup başvuruda ürünün biyobenzer olduğunun belirtilmesi gerekmektedir.
Türkiye’de sağlık alanındaki pek çok mevzuat gibi, biyobenzerlere ilişkin düzenlemeler de Avrupa Birliği regülasyonları ile paralellik arz etmektedir. Bu kapsamda 2001/83/AT sayılı Beşeri Tıbbi Ürünler Hakkındaki Avrupa Parlamentosu ve Konsey Direktifi’ne uyumlu olarak hazırlanan ve 2005 yılında yürürlüğe giren Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği’ne dayanılarak, Benzer Biyolojik Tıbbi Farmasötik Ürünlerin Ruhsatlandırma Kriterlerini belirlemek amacıyla 2008 yılında Biyobenzer Tıbbi Ürünlere İlişkin Kılavuz (“Kılavuz”) yürürlüğe konulmuştur. Bu kapsamda biyobenzer ilaç, ruhsatlı biyolojik referans bir ilaca benzerlik gösteren ilaçlara verilen ad olarak tanımlanmakta, referans biyolojik tıbbi ürünün ise idari, kalite, ve klinik verileri de kapsayacak şekilde tam başvurusu ülkemiz dışında ilgili otoriteye yapılmış ruhsatlı bir ürün olması aranmaktadır.
Açıklandığı üzere FDA ve EMA, klinik olmayan in vivo çalışmaların ve klinik çalışmaların kapsamını azaltmak maksadıyla biyobenzerler bakımından aşamalı bir yaklaşım benimsemektedir. Ancak, fizyokimyasal, biyolojik ve klinik olmayan in vitro verileri içeren önceki adım(lar)da elde edilen kanıtların yeterli düzeyde olması aranmaktadır. Karşılaştırılabilirlik bakımından CFDA de önce analitik, sonra klinik olmayan ve ardından klinik çalışmalar yapılmasını öngören aşamalı bir yaklaşım benimsenmektedir. Kılavuz uyarınca, biyolojik ve biyoteknolojik ürünlerin karmaşıklığına bağlı olarak biyobenzer ürünlerin ruhsatlandırılmasında jenerik yaklaşımın uygun olmadığı, kalite güvenlik ve etkinlik açısından karşılaştırabilirlik çalışmaları yapılması gerektiği belirtilmiş olup bu kapsamda ruhsat dosyası hazırlanmasında farklılıkları ortaya koyan tabloya yer verilmiştir.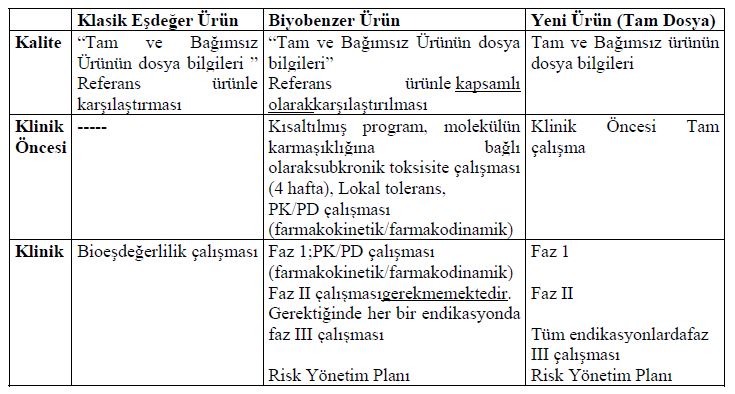
Çin regülasyonlarında biyobenzerlerin isimlendirilmesine ilişkin bir düzenlemeye yer verilememekle birlikte Amerika Birleşik Devletleri ve Avrupa Birliği’nde ise hastaların rekabetçi ve güvenli bir biyobenzer pazarından yararlanmalarını sağlamak maksadıyla isimlendirme konusunda gereklilikler regüledir. Örneğin Avrupa Birliği regülasyonları kapsamında tüm biyolojik ilaçların klinik kullanım sırasında ve tedarik zincirinin tüm seviyelerinde ürün ve parti izlenebilirliğini sağlamak adına, biyobenzerlerin etken madde adı ile birlikte bir de ticari veya marka adı kullanılarak isimlendirilmesi gerekmektedir. Kılavuz uyarınca da, biyobenzer ilaçlar sadece ticari ismi, görünüş ve ambalajlama özellikleri açısından biyolojik referans ilaçlardan farklılık göstermektedir.
Türkiye İlaç ve Tıbbi Cihaz Kurumu’nca (TİTCK) biyobenzerlerin ruhsatlandırılmasında EMA veya FDA uygulamalarına benzer şekilde aşamalı bir yöntem izlenmektedir (Görsel 6). İlaveten, 2001/83/AT sayılı beşeri tıbbi ürünler hakkındaki Avrupa Parlamentosu ve Konsey Direktifi ile 726/2004/AT sayılı beşeri tıbbi ürünler için koşullu ruhsat hakkındaki Avrupa Parlamentosu ve Konsey Tüzüğü’ne uyum çalışmaları kapsamında TİTCK resmi internet sitesinde taslak Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği ve taslak Biyobenzer Tıbbi Ürünler Hakkında Kılavuz yayımlanmış olup bu mevzuat henüz yürürlüğe girmemiştir.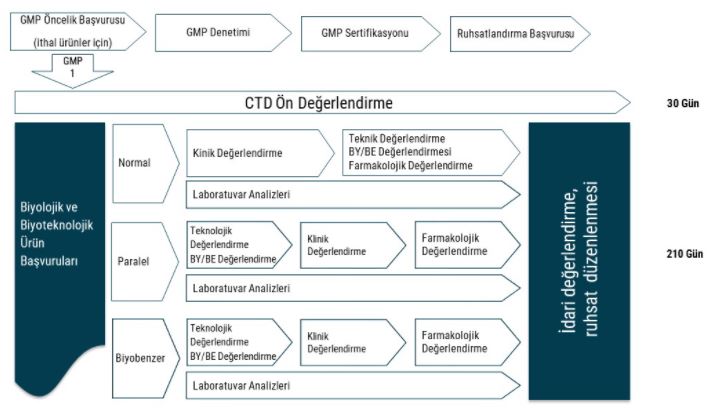
Görüldüğü üzere düzenleyici otoritelerin çoğu biyobenzer ilaçların ruhsatlandırılması için kısaltılmış yöntemleri tercih etse de biyobenzerleri piyasaya sunmaya hazır hale getirmenin kendine has zorlukları bulunduğu ortadadır. İlaveten, referans ürünle değiştirilebilirlik ve karşılaştırılabilirlik bakımından detaylı regülasyon ve kriterler nedeniyle biyobenzerlerin referans ürünleriyle terapötik eşdeğerler olarak değil, terapötik alternatifler olarak rekabet etmesi gerekmektedir.
Av. Şafak HERDEM
HERDEM Avukatlık Bürosu kurucu ortağı olan Şafak Herdem, yurtiçinde ve yurtdışında özellikle savunma sanayi, yenilikçi teknolojiler ve uluslararası ticaret alanlarında hukuki danışmanlık hizmetleri vermekle, Bilgi Üniversitesi’nde de öğretim görevlisidir. Uluslararası Barolar Birliği ve Amerikan Barolar Birliği üyelikleri de bulunan Herdem’in aynı zamanda Savunma Sanayi Hukuku adında bir de eseri bulunmaktadır.
Başvurular:
Chen Y., Lehmann A., & da Silva, J. S. What’s next for biosimilars in emerging markets? Erişim tarihi 03.01.2021. McKinsey&Company: https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/whats-next-for-biosimilars-in-emerging-markets
WHO. Expert Committee on Biological Standardization Sixtieth Report Annex 2: Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Erişim tarihi 03.01.2021. https://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=1&ua=1
EMA. Guideline on similar biological medicinal products (CHMP/437/04 Rev 1.) Erişim tarihi 03.01.2021. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
EMA. (2019) Biosimilars in the EU Information Guide for Healthcare Professionals.
İlaç ve Eczacılık Genel Müdürlüğü. Biyobenzer tıbbi ürünlere ilişkin kılavuz 07.08.2008.
Kumar R, Sigala S, Malgarini RB, Pimpinella G, Pani L, et al. (2015) Biosimilars: Regulatory Status and Implications across the World. J Pharmacovigilance S3: 002.
FDA. Biological Product Definitions.
FDA. Biosimilar Product Regulatory Review and Approval.
TITCK. Beşeri Tıbbi Ürünler Ruhsatlandırma Yönetmeliği Taslağı. Erişim tarihi 03.01.2021. https://www.titck.gov.tr/mevzuat/beseri-tibbi-urunler-ruhsatlandirma-yonetmeligi-taslagi-27122018172955
TITCK. Biyobenzer Tıbbi Ürünler Hakkında Kılavuzu Taslağı. Erişim tarihi 03.01.2021. https://www.titck.gov.tr/mevzuat/biyobenzer-tibbi-urunler-hakkinda-kilavuzu-taslagi-27122018173016
EPO. Patent Index 2019 Sataistics at a Glance. Erişim tarihi 09.01.2021. http://documents.epo.org/projects/babylon/eponet.nsf/0/BC45C92E5C077B10C1258527004E95C0/$File/Patent_Index_2019_statistics_at_a_glance_en.pdf
FDA. Biosimilars. Erişim tarihi 03.01.2021. https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars
TITCK. İlaç Ruhsatlandırma. Erişim tarihi 03.01.2021. https://www.titck.gov.tr/faaliyetalanlari/ilac/ilac-ruhsatlandirma

